�l(f��)���r�g�����ٷ��Ӌ��C(j��)�QՓ���g�[��1��
ժ Ҫ�� ժҪ ���^ȥ��10����, �Ի���M�W(xu��)���t(y��)�W(xu��)�z���W(xu��)����(j��ng)��Ϣ�W(xu��)�Ȟ�����������ƌW(xu��)���о��I(l��ng)��, ��ǰ��δ�е����Lڅ��, �e���˺����Ĕ�(sh��)��(j��)��Ϣ. �@Щ��(sh��)��(j��)��͏�(f��)�s����(sh��)������, �����N(y��n)���ărֵ���Dz��ɹ���. ͨ�^���y(t��ng)��̎���ֶ�, �y�����庣��ԭʼ��(sh��)��(j��)���e�C��(f��)�s���P(gu��n)(li��n)��Ϣ. ��
����ժҪ ���^ȥ��10����, �Ի���M�W(xu��)���t(y��)�W(xu��)�z���W(xu��)����(j��ng)��Ϣ�W(xu��)�Ȟ�����������ƌW(xu��)���о��I(l��ng)��, ��ǰ��δ�е����Lڅ��, �e���˺����Ĕ�(sh��)��(j��)��Ϣ. �@Щ��(sh��)��(j��)��͏�(f��)�s����(sh��)������, �����N(y��n)���ărֵ���Dz��ɹ���. ͨ�^���y(t��ng)��̎���ֶ�, �y�����庣��ԭʼ��(sh��)��(j��)���e�C��(f��)�s���P(gu��n)(li��n)��Ϣ. ��ᘌ������(sh��)��(j��)�Ŀ�ҕ���о�, �������ڿ����ˆT����(f��)�s��(sh��)��(j��)�M(j��n)�ж�Ƕ��^�첢�@ȡ��Ч��Ϣ. ���(sh��)��(j��)��Խ��, ��(f��)�s��Խ��, ��ҕ����������Ч��Ϣ�ھ���l(f��)�]�����þ�Խ��. ����ͨ�^���e��������C(j��)��(g��u)���ĬF(xi��n)��Ĕ�(sh��)��(j��)Ҏ(gu��)ģ�͔�(sh��)��(j��)���L����, �f�������о��I(l��ng)�����M(j��n)���(sh��)��(j��)�r��, Ȼ�������(sh��)��(j��)�ĽM����������ҕ�������c���������(sh��)��(j��)��ҕ������Ҫ�Ժͱ�Ҫ��. ���Ŀ��Y(ji��)�������ƌW(xu��)�о��I(l��ng)���в�ͬ��������(sh��)��(j��)�Ŀ�ҕ���о��M(j��n)չ, ���ӑՓ��Ŀǰ�����(sh��)��(j��)��ҕ�������R������(zh��n), ��������ܵĽ�Q����.
�����P(gu��n)�I�~ ��(sh��)��(j��)������Ϣ�W(xu��)��ҕ��
����2005���ԁ�, �S����ͨ���y���g(sh��)�IJ����M(j��n)�����V����(y��ng)��, �����ƌW(xu��)�����˴�(sh��)��(j��)�r��. �Ի���M�ƌW(xu��)�������t(y��)�W(xu��)�I(l��ng)�������������ƌW(xu��)�о��a(ch��n)���ͷe���˺����Ĕ�(sh��)��(j��)��Ϣ: �W��������Ϣ�W(xu��)�о��� (European Bioinformatics Institute, EBI)Ŀǰ�惦�ˌ���20 PB�Ĕ�(sh��)��(j��), ���л���M��(sh��)��(j��)�sռ2 PB, �@һ��(sh��)���S����һ���y���g(sh��)�IJ���l(f��)չÿ��ɱ����L[1]; ��ͨ���y��(sh��)��(j��)��(Sequence Read Archive, SRA)���������������\�g(sh��)��Ϣ����(National Center for Biotechnology Information, NCBI)����Ҫ�ĸ�ͨ����(sh��)��(j��)�惦��, Ŀǰ�惦�Ĕ�(sh��)��(j��)�������^��3 PB, ����l(f��)���Ĕ�(sh��)��(j��)���_(d��)��1640 TB[2]; ����, ��(d��ng)ǰ���������Ļ���(sh��)��(j��)�a(ch��n)���C(j��)��(g��u)——�A������о�Ժ(Beijing Genomics Institute, BGI)ÿ��a(ch��n)�������ˡ�ֲ�����������ڃ�(n��i)�ļs6 TB����M��(sh��)��(j��)[1].
�������H�ϵĶ��������о��Ŀ�a(ch��n)����ʷ�oǰ��Ҏ(gu��)ģ�����(sh��)��(j��). �������g���ȫ���z����Ϣ, �����ƌW(xu��)����1985����������������MӋ��(Human Genome Project, HGP)[3], �@һӋ�����H���w�� 99.99%�������M, ���x�����w�����ܴa��“����֮��”, �����Ƅ��������ƌW(xu��)�����\�g(sh��)�Ļ��A(ch��)���о�, ���M(j��n)��һϵ�пƌW(xu��)���g(sh��)�Įa(ch��n)���Ͱl(f��)չ; 2004��, ���ˌ�����һ��DNA�о����g(sh��)��������{(di��o)��������ȫ����Mˮƽ���о��đ�(y��ng)��, “DNAԪ���ٿ�ȫ��” Ӌ��(Encyclopedia of DNA Elements, ENCODE)����, �@һӋ����ʹ����32�����ЙC(j��)��(g��u)��442���о��ˆT�@ȡ�������˳��^15 TB��ԭʼ��(sh��)��(j��)[4]; ��2005�������, �����������Ұ��Y�о���(National Cancer Institute, NCI)���������������M�о���(National Human Genome Research Institute, NHGRI)��ͬ�l(f��)��İ��Y����M�D�VӋ��(The Cancer Genome Atlas, TCGA)[5], ͨ�^��������M���g(sh��)ƽ�_�������@ȡ���^ 800 TB��(sh��)��(j��)���ęn�Y��, ���\�ࡢ�ί����A(y��)�����Y�����˶،��Ļ��A(ch��); 2010��, �С�Ӣ���¡����ȇ���ͬ�����ˇ��Hǧ�˻���MӋ��(1000 Genomes Project), �����ֹ�a(ch��n)���Ĕ�(sh��)��(j��)���_(d��)��50 TB, ���а�������ȫ��27����Ⱥ��2500���˵�ȫ������M��Ϣ[6].
������(d��ng)��y���g(sh��)���M(j��n)���ٶ�֮��, ���h(yu��n)��Ӌ��C(j��)�I(l��ng)�����Ħ������[7](�r��׃�r, �����·������ÿ18��������һ��). ��1990�ꆢ�ӵ������MӋ����, �����W���С��յȶ������Һ͵^(q��)���^200���ƌW(xu��)��, Ͷ���˳��^10��ĕr�g�ͼs30�|��Ԫ��������ȫ����M�Ĝy��; ���F(xi��n)��, �H��һ������ҵĔ�(sh��)���о��ˆT, �Ϳ��ڔ�(sh��)�܃�(n��i)������ȫ����M�y��, ��ԇ���ɱ��t�ɿ�����1000��Ԫ֮��(n��i). ��˾���M(j��n)��, ���H�o�����ƌW(xu��)���о������˾�ęC(j��)��, �ڴ˻��A(ch��)�������Ч̎���ͷ����@Щ�y��(sh��)��(j��), Ҳ�o���I(l��ng)���(n��i)���о��ˆT�����˾������(zh��n).
������DNA���Д�(sh��)��(j��)������ĺ�����(sh��)��(j��)�ǘ�(g��u)�������ƌW(xu��)�о�����Ҫ�M�ɲ���, ͨ�^��(y��ng)��������Ϣ�W(xu��)���g(sh��)�M(j��n)�д�(sh��)��(j��)�о�, �����[���ڴ�(sh��)��(j��)�������W(xu��)֪�R�ɞ鮔(d��ng)ǰ���\�g(sh��)�l(f��)չ����������. ���y(t��ng)�Ļ����ı��Ĕ�(sh��)��(j��)̎����չʾģʽ�ѽ�(j��ng)��(y��n)���Ƽs�ˌ��������ƌW(xu��)��(sh��)��(j��)�Ľ��x. ���ڿ�ҕ�����g(sh��)����Ϣ�ھ�ɞ�һ�N����Ľ�Q;��. ��ҕ���nj����ィ������ģ�ͻ�������D���һ���^��1). ͨ�^��ҕ��, ����ķ�̖��Ϣ�����D(zhu��n)������������ĈD���ģ��, ���⽻��ʽ��ʹ�����S�о��ˆT�IJ�ͬ�Ŀ�ҕ���Ƕȁ�̽���[���ڴ�(sh��)��(j��)��IJ�ͬģʽ���P(gu��n)(li��n). ��ҕ�����Џ�(qi��ng)��Č���(f��)�s��(sh��)��(j��)�D(zhu��n)�����������Ϣ������. ���(sh��)��(j��)��(f��)�s��������T�����c�Q���˿�ҕ������Ч���������(sh��)��(j��)�IJ��ɻ�ȱ���ֶ�. �����(sh��)��(j��)��ҕ�������ڬF(xi��n)�е�Ӌ�㼼�g(sh��), ��һ���r�g��(n��i)�a(ch��n)��ҕ�X���F(xi��n)ģ��, ���ڴ˻��A(ch��)�ϱM���ܵ�����(qi��ng)������, �Ķ��ӏ�(qi��ng)�Ñ��w��Լ������(sh��)��(j��)�����Y(ji��)�����J(r��n)֪����.
����1 �����(sh��)��(j��)����������Դ
���������(sh��)��(j��)���˾��Ђ��y(t��ng)��(sh��)��(j��)4“V”�����c, ����(sh��)��(j��)����(Volume)����(sh��)��(j��)̎���ٶȿ�(Velocity)����(sh��)��(j��)Դ��׃(Variety)���N(y��n)���rֵ(Value)��[8], ߀���������еĔ�(sh��)��(j��)��(f��)�s��(Complexity)[9]. ������W(xu��)�����, ��(f��)�s�̶Ȍ������I(l��ng)��a(ch��n)���Ĵ�Ҏ(gu��)ģ��(sh��)��(j��)�c�����ƌW(xu��)�I(l��ng)��Įa(ch��n)���^(q��)���_��. �ڸ���������, ��(sh��)��(j��)���������ĽY(ji��)��(g��u)��ע�, ������W(xu��)��(sh��)��(j��)Ŀǰ���v�s�y�������ؽM������. ���˺��εĻ���M�y����, ����W(xu��)�ҕ�ۙ�S�ͬ�ļ�(x��)���ͷ��ӳɷ�, ԇ�Dʹ�ø��N�ֶ�Ū�����а����ď�(f��)�s�P(gu��n)ϵ. ����, �������(sh��)��(j��)��(j��ng)�����Բ�ͬ�Č����͙C(j��)��(g��u), ʹ���˲��M��ͬ�ą���(sh��)��(bi��o)��(zh��n), �a(ch��n)���Ĕ�(sh��)��(j��)����S������, ��(d��o)���@Щ��(sh��)��(j��)���ܲ��ò�ͬ�Ĵ惦�Y(ji��)��(g��u)(��narrowPeak, BED, SAM ��), ᘌ���ͬ���о�����(��������С������|(zh��)�����P(gu��n)ϵ����Ⱥ������), ��Դ�ڲ�ͬ������(��y���t(y��)��ӛ䛵�). ��ͬ���ą���(sh��)��(bi��o)��(zh��n)���خ��ļ�(x��)���M������Լ��o���Y(ji��)��(g��u)���惦��ˎ��̎���^�̵��T�����ض�����������(sh��)��(j��)��(f��)�s�Ե�ԭ��. ���(sh��)��(j��)��ҕ���ĺ��ľ���������Ч���㷨�����@Щ��(sh��)��(j��)�ď�(f��)�s��, �Ķ��������[��������W(xu��)Ҏ(gu��)��������չʾ�o�Ñ�, ���������D(zhu��n)�Q�@Щ��(f��)�s��(sh��)��(j��)�ĸ�ʽ, �t�ǔ�(sh��)��(j��)��ҕ���O(sh��)Ӌ�ĵ�һ��, �����Ԕ�(sh��)��(j��)��Դ�����˽����(sh��)��(j��)��(f��)�s���ӵĸ�ʽ����.
��������, �y���g(sh��)���w�ٰl(f��)չ�������I(l��ng)���ṩ�˔�(sh��)Ŀ����Č��F�YԴ. Ŀǰ�ڶ����y���g(sh��)���V������, �ڶ����y��a(ch��n)����(sh��)���fӋ�Ķ�����, ����ƴ���㷨���@Щ��������ȫ����M������(n��i)�M�b����, �Ķ��M(j��n)���M(j��n)һ���Ĕ�(sh��)��(j��)��������[10]. �����ֹ, ���d�Ćμ�(x��)���y���g(sh��)һֱ���J(r��n)�������ֵ���P(gu��n)ע�Ĝy���g(sh��), ���y(t��ng)�Ĝy�������˼�(x��)���g�IJ��, �õ��ĽY(ji��)���H�H��һȺ��(x��)����̖��ƽ��ֵ, �����چμ�(x��)��ˮƽ��ȫ����M�M(j��n)�ДU(ku��)���c�y��Ćμ�(x��)���y���g(sh��), ���H�ڻ�����_(d��)������y������(zh��n), �����܉�z�y�����_(d��)���^�͵Ļ��Ǿ��aRNA, ��˾��кܴ�ă�(y��u)�ݼ��l(f��)չ���g[11]. ����֮��, �μ�(x��)�� RNA�y��(single-cell RNA-seq)ʹۙ����(x��)�����D(zhu��n)䛽M�ɞ���� [12], Ⱦɫ�|(zh��)���߹�����y�� (ChIP-seq)[13]�Ȍ��g(sh��)������֧���ˌ�����M��(sh��)��(j��)�Ĺ�����ע�. �@Щ��ͨ���Ĝy���g(sh��), ���о��߰l(f��)�F(xi��n)�c�������P(gu��n)�Ļ�����׃�����о�ij�����͵������D(zhu��n)䛽M��ijһ�l���µļ�����B(t��i)�Լ���DNA�ϵ����|(zh��)�Y(ji��)��λ�c�M(j��n)�ж�λ�ȹ����ṩ�˱����c֧��, Ȼ���S����(sh��)��(j��)Ҏ(gu��)ģ������, �y��(sh��)��(j��)��̎���ͷ�����u�ɞ�ƿ�i.
�������, ����оƬ���g(sh��)��ʹ�����^ȥ�Ĕ�(sh��)���Юa(ch��n)��������Ĕ�(sh��)��(j��)�YԴ. ���ˌ��F(xi��n)������M������(x��)���������|(zh��)������ȽM���и����Ĵ�����Ϣ�M(j��n)�п��ٜ�(zh��n)�_�ęz�y, �о��ˆT�ڹ��wоƬ���昋(g��u)�����͵����ﻯ�W(xu��)����ϵ�y(t��ng). ��(d��ng)ǰ������оƬ��Ҫ�֞����оƬ������оƬ�ɷN���[14]. ���y(t��ng)�����o�B(t��i)���s�����g(sh��)����A(ch��)�����оƬ��Ҫ�л���оƬ(DNA Microarray)������оƬ(Protein Chip)��оƬ����� (Lab-on-a-chip)����ʽ[15]. ����, ����оƬҲ��DNA ���ܶ��c��s�����g(sh��), �Ժ���̽ᘻ��a(b��)�s�����g(sh��)����A(ch��)������, ������DNA���Мy������_(d��)��������������Լ�������B(t��i)�Է������о�Ŀ��; ����оƬ����(j��)�����|(zh��)���Ӻ��������ӵ�����ö���(g��u)��; ��оƬ����Ҍ��������̼��s���γ��͵ķ���ϵ�y(t��ng). оƬ�c������ӷ���(y��ng)���a(ch��n)������̖��Ҫ������оƬ����x, ��ͨ�^���P(gu��n)ܛ�������ɼ����ĸ�����(y��ng)�c�ğɹ⏊(qi��ng)����̖������λ����Ϣ���γɵĈD����@ȡ���P(gu��n)��������Ϣ. ����оƬ�����w���Ƽ��g(sh��)����A(ch��), ��Ҫ��ë��(x��)���ӾоƬ��PCR����(y��ng)оƬ����ʽ[15]. �����, ����оƬ���g(sh��)�ڻ�����_(d��)ˮƽ�z�y�������\�ࡢˎ��Y�x�����w���t(y��)���R���������\����ί��������л���l(f��)�F(xi��n)�Լ������ܴ_�J(r��n)���t(y��)�W(xu��)�c����W(xu��)�I(l��ng)��õ��V���đ�(y��ng)��.
�����ٴ�, �����|(zh��)�V�������ƌW(xu��)���о������˾��ؕ�I(xi��n), ���H���J(r��n)���Ǵ�Ҏ(gu��)ģ����ͨ���b����ʮ�f���������������ӽY(ji��)��(g��u)�����x����, ���Ҍ����о�����-���ȴ����֮�g������á����g������Լ�������_(d��)ˮƽ��׃�������ܴ�Ď���. �|(zh��)�V����Ҫԭ�����Ȍ���Ʒ׃?y��u)��B(t��i)���x�ӻ����, �ٰ����|(zh��)�ɱ�(m/z)�M(j��n)�з��x, �Ķ��ɹ��@ȡ��Ʒ���|(zh��)�����������Y(ji��)��(g��u)����Ϣ[16]. �ګ@ȡʹ���V�D�����б�����ʾ�Ĝy���Y(ji��)����, ��Ҫ�M(j��n)���M(j��n)һ���Ĕ�(sh��)��(j��)����. �����b�������|(zh��)�ķ���, Ŀǰ���õ����|(zh��)���y�b���� (Peptide Mass Fingerprinting)�������|(zh��)�V�Ĕ�(sh��)��(j��)�������b����(MS/MS Database Searching)���ֶ�[17]. �|(zh��)�V�������g(sh��)���Q�������|(zh��)�M�ĺ��ļ��g(sh��), ����� Nature�Ϲ�����������|(zh��)�M�݈D���ǻ���16857���|(zh��)�V�������Y(ji��)��������[18]. ���|(zh��)�o���������-�w�Еr�g�|(zh��)�Vϵ�y(t��ng)(VITEK®MS)��������FDA����(zh��n)���ׂ����ڙz�y�������|(zh��)�V�z�yϵ�y(t��ng), �����ڽ�ĸ�����²���(x��)���R�������b��, �@Ҳ�ǵ�һ�N���ڔ�(sh��)��犃�(n��i)�z�y�²�������t(y��)����е[19].
��������, ͨ�^���N���M(j��n)�ֶΫ@ȡ���c�������P(gu��n)�ĈDƬӰ���Y��Ҳ�����S������. �����w��(n��i)�����������|(zh��)��RNA�Լ�DNA�ȷN������������. �S���@�R�������ȸ߾���˃x�����g(sh��)�IJ���l(f��)չ, �ƌW(xu��)�҂����H�܉�ͨ�^�͜�����@�Rֱ���^�쵽�����|(zh��)���������Ӿ���(x��)��ԭ�ӵĽM���Y(ji��)��(g��u), ������u����ֱ���^�yӛ䛵����w�M�������������ڕr�g�����g�S���ϵĽY(ji��)��(g��u)׃���������g������õĄӑB(t��i)����. Ŀǰ, ����˹̹����W(xu��)�о��ˆT���� “�̓�(n��i)�Q�R”��������(d��o)���ѽ�(j��ng)���F(xi��n)���ڲ��ƉĻ��w���^��M������r��, �L�r�g�،����w���X��(j��ng)Ԫ�M(j��n)���^�y[20]; ������W(xu��)�_�l(f��)��“���������ܼ�����ɢ�����”���g(sh��)�ɹ����خ��Ԙ�(bi��o)ӛ�˻(x��)����֬����ǡ������|(zh��)����ȳɷ�[21]; �����~�s��Ȫ�ی���Ҍ����Ә�(bi��o)ӛ�ֶ��c�@�R���g(sh��)��Y(ji��)��, �������˵�һ�����w�����w��(n��i)�[����(x��)����ӵ�Ӱ��ӛ䛹���[22]. ͨ�^�@Щ���¼��g(sh��)�ֶ�, �ƌW(xu��)�҂��������еõ����м�(x��)�����M���е����|(zh��)�͏�(f��)��������P(gu��n)λ��, Ū�����w���ЙC(j��)��śr. ���, Խ��Խ��ǽY(ji��)��(g��u)���ĈDƬӰ��(sh��)��(j��)ؽ�����������ϡ�������չʾ.
�������, �R����(sh��)��(j��)Ҳ��һ�����ɺ��ԵĔ�(sh��)��(j��)��Դ. �H�`�����Ї����t(y��)�ƌW(xu��)Ժ�ďV���T�t(y��)Ժÿ��a(ch��n)���Ĕ�(sh��)��(j��)�����_(d��)��70 TB2), �����ȫ�����R����(sh��)��(j��)��������һ��, �䔵(sh��)��(j��)Ҏ(gu��)ģ���Dz��ɹ���. �F(xi��n)�е��R���t(y��)�W(xu��)��(sh��)��(j��)������Ӳ��v���t(y��)�W(xu��)Ӱ���Y(ji��)���Լ������z�顢������Ƭ�z�������W(xu��)��Ϣ��, �@Щ�R����Ϣ�������ӡ����ࡢ������, ���������漰�����[˽����˾����_ͻ�Ȇ��}, ��֮��Щ��(sh��)��(j��)֮�g�y���P(gu��n)(li��n), ��ɘ�(bi��o)��(zh��n)����ʩ�����y. �@�N�Y(ji��)��(g��u)���c�ǽY(ji��)��(g��u)����ʽ��������c, ʹ���R����(sh��)��(j��)����������׃�î������y[23]. �����ھ��@Щ�t(y��)����(sh��)��(j��)�Н��ڵărֵ, һЩ�R���Ϳ��ЙC(j��)��(g��u)�����t(y��)����(sh��)��(j��)�M(j��n)������, ��(g��u)���R��ԇ(sh��)��(j��)�Ĺ����ͷ���ƽ�_. �����ĸ����t(y��)Ժͨ�^�R��������Ϣ����ϵ�y(t��ng)�����`��(sh��)��(j��)����Ҏ(gu��)��������(sh��)�ֻ�, �����Ĕ�(sh��)��(j��)ͨ�^�����D(zhu��n)�Q���^��, ���M(j��n)һ����(y��ng)���ڲ�ԃ�z�����y(t��ng)Ӌ�����͔�(sh��)��(j��)�ھ���, �Դ˫@ȡ�µ�֪�R, �Ķ�������Ч�،��R�����`�M(j��n)��ָ��(d��o)2). �����R���[���W(xu��)��(American Society of ClinicalOncology, ASCO)���µ�“CancerLinQ”���S�о��ˆT�M(j��n)�롢�L���ͷ����������Y���ߵIJ���[24]; ���͵�����\���I(l��ng)��Ҳ����Ϣ�����ṩ�˘O��ı���. �������R����(sh��)��(j��)���������Ì���������ڿ����ˆT���t(y��)�W(xu��)���Ҍ���Ҏ(gu��)ģ��������Ⱥ�w�ί���r�M(j��n)�з���, �Ķ��鹥�����y�s�Y�ṩ���C(j��).
�������P(gu��n)֪�R���]���Ք�(sh��)��(j��)��ҕ��Փ�ĵ��ڿ�
�������������ׂ���Ҫ�������(sh��)��(j��)��Դ����, ���͵ļ��g(sh��)�ֶβ���ؕ�I(xi��n)�����F���YԴ��(sh��)��(j��), �������µ���ʽ�ɹ⼼�g(sh��)[25]���Ԍ��F(xi��n)���١���(zh��n)�_����ͨ���،��[����(bi��o)־���M(j��n)�Йz�y, ���ⲻͬ��͵ăx���O(sh��)��Ҳ�������I(l��ng)���ṩ�˲����Ѓrֵ�Ĕ�(sh��)��(j��). �S���Ĕ�(sh��)��(j��)��Դ�@ʾ�����(sh��)��(j��)���H��(sh��)��(j��)Ҏ(gu��)ģ����, ��͏�(f��)�s��׃, ���������w���g�ϽY(ji��)��(g��u)��λ���S�r�g����׃�Q���Ƅ�. ��Q�@Щ��(sh��)��(j��)�Ĵ惦ֻ����������΄�(w��), ������Ҫ����ʹ���@Щ��(sh��)��(j��). ͬ��, �������(sh��)��(j��)�M(j��n)�п�ҕ���Ǟ��˸��ӳ�ֵ��ھ����(sh��)��(j��)�Н��ڵărֵ, ������O(sh��)Ӌ��ҕ�����ߕr����܉��Ԕ�(sh��)��(j��)��Դ������(j��), �Ĕ�(sh��)��(j��)Ҏ(gu��)ģ����(f��)�s�ȡ����g�Ժ͕r�g׃�Q���@4������ᘌ�Ŀ��(bi��o)��(sh��)��(j��)�M(j��n)�п��], ��ʮ�������ڏĔ�(sh��)��(j��)�Ы@ȡ��Ч��Ϣ.
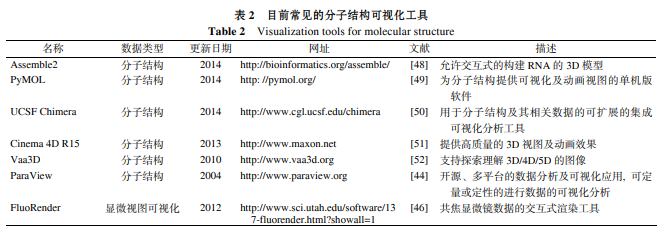
����2 �����(sh��)��(j��)��ҕ����ͼ��F(xi��n)��
������ҕ�������(sh��)��(j��)�ķ������P(gu��n)��Ҫ, �����(sh��)��(j��)�����ԁ���, һ����r�H�{���ֺ��y����������д��ڵď�(f��)�s�P(gu��n)ϵ. ��ҕ�����H�����Á��M(j��n)������չʾ, ���ǔ�(sh��)��(j��)�����ĵ�һ����(zh��n)��, �����(sh��)��(j��)�M(j��n)�����õ�ֱ�^��������չʾ���Խ�ʾ����(sh��)��(j��)��(n��i)�ڵ��e�C��(f��)�s���P(gu��n)(li��n)��r, ���@һ�c�������������y�c��ҕ�����ᲢՓ. ����ε�Excel��ӱ���Google �ęn��R, Pandas�Ƚy(t��ng)Ӌ���̼ܘ�(g��u), �ٵ�D3.js, Prefuse �ȿ�ҕ�������, �@Щͨ�Ô�(sh��)��(j��)��ҕ����̎�����߶����Ԟ锵(sh��)��(j��)��������Ϣ�ھ��ṩ�ܺõ�Ӌ��C(j��)�ֶ�. ����ᘌ��ڲ�ͬ�Ĕ�(sh��)��(j��)��ͺ�Ŀ��, �����I(l��ng)��ӿ�F(xi��n)��һ�����_Դ����(y��u)��Ŀ�ҕ������(�D1), �@Щᘌ������о��ˆT�_�l(f��)�Ĺ�����������, �����(sh��)��(j��)�Ŀ��ٷ����ṩ�˱���.
����2.1 �y��(sh��)��(j��)
�����y���g(sh��)������оƬ�ṩ�˴����������w�е� DNA, RNA, �����|(zh��)�ȴ���ӵ��S����һ�������YԴ, �F(xi��n)�еĻ���M�g�[������(j��)��ͬ�������@Щ�����M(j��n)���ˏļ�(x��)��(ji��)�����^��չʾ. �Ԯ�(d��ng)ǰ��鳣�õ� UCSC Genome Browser[26]����, ��֧�ֿ��Ա��Ȍ�������M�ϵ��κΔ�(sh��)��(j��)���, ���D���ڷ���(w��)������Ⱦ��Ƕ��W(w��ng)���. �����ڻ���M��(sh��)��(j��)��չʾģʽ�w�F(xi��n)�˴��F(xi��n)��g�[�����е����c: (1) ��Ⱦɫ�wλ�Þ������Ļ���M��(sh��)��(j��)ҕ�D; (2) �ԅ�������M���(bi��o)��(zh��n)�ṩλ������(bi��o)�S; (3) ����trackչʾ; (4) ���õĽ����ԺͿɶ�����, �ɸ���(j��)�Ñ������M(j��n)���b�d���[�ؔ�(sh��)��(j��)��(n��i)��. �����@Щչʾ������, ��ͬ�Ļ���M�g�[��Ҳ�����Լ����صĹ���. GenomeView[27]�ṩעጾ���, ����չʾ��ע���Ϣ, �M(j��n)�ж����бȌ���������ƥ�䡢�����бȌ��Լ��������Ա��@ʾ�ă�(n��i)��; ����̽�����ͼ��ɔ�(sh��)��(j��)���Ŀ�ҕ������ (Integrative Genomics Viewer, IGV)[28]��֧�ֶ�N��(sh��)��(j��)��͵Ľ���չʾ, �����y�����бȌ���������_(d��)��(sh��)��(j��)�Ϳ�ؐ��(sh��)����(�D1(e))��.
�������ڲ�ͬ�ĽM���D(zhu��n)䛽M�ı��_(d��)����������ڽy(t��ng)Ӌ�ֶ��M(j��n)�о��, ����Ҫʹ�ß�Dʹ��Y(ji��)���ʬF(xi��n)ֱ�^��չʾ, �����Խ��, ��õ��IJ�ͬ���_(d��)ģʽ߀���M(j��n)һ�����չ��ܸ����̶��M(j��n)�з���ԈD�λ���ʽ��ʾ���O(sh��)�z�ĽY(ji��)��, ��Gitools[29]������Ĵ���߲����˟�D����ʽ������M��(sh��)��(j��)�M(j��n)�м��ɻ�������չʾ, �˹���ͨ�^����KEGG, Biomart�����(sh��)��(j��)���_(d��)�������֪�R������, �ṩ�������������P(gu��n)�Է����Լ��@����Ӌ����S���ķ����ֶ�, ͨ�^���������^�V���Ƅӡ��ۼ�����������ҕ������עጵȹ������Sʹ���߽����Եط����Ϳ�ҕ����S��(sh��)��(j��).
��������, �y��(sh��)��(j��)�Ŀ�ҕ�����ܕ�����(sh��)��(j��)�������ھ������Q���Ե�����. ����, �κ�������B(t��i)�� (SNP)������ȱʧ��(bi��o)ӛ(InDel)�Լ�����M�Y(ji��)��(g��u)׃����һ���������H���P(gu��n)ע�ă�(n��i)��, ���������c��(f��)�s�����İl(f��)���l(f��)չ���������P(gu��n)ϵ. ����, ����M�Y(ji��)��(g��u)׃���������롢�h�������á���λ����(f��)���Լ���ؐ��(sh��)׃���Ȳ�ͬ�����, ÿ�N���ʹ����M�a(ch��n)����ͬ�ĽY(ji��)��(g��u)��׃. ���ڸ�Y(ji��)��(g��u)׃���ď�(f��)�s��, �Լ�����������M�Y(ji��)��(g��u)���е��؏�(f��)��������, ��(d��o)�H�{�F(xi��n)�е��㷨���y��ȫ���_�ؙz�y��ÿ�N��͵�׃��. �e��, �Y(ji��)��(g��u)׃����������������е��e�`��λ, �M(j��n)����(d��o)��С�߶ȵĶ��B(t��i)���A(y��)�y�e�`, ���ͨ�^�ṩ��ҕ�����߁������о����M(j��n)���˹��Д��ڽY(ji��)��(g��u)׃���ęz�y���R�e��׃�ò��ɻ�ȱ[30]. Ŀǰ�����T���������չʾ��̽���Y(ji��)��(g��u)׃���Ŀ�ҕ������, ������\���ڸ��N����ϵ�y(t��ng)�ϵ�ᘌ��Y(ji��)��(g��u)׃���ļ���ܛ��inGAP-sv[30], ���H�܉����^�͵ļ���Ը��ʙz�y����(f��)�s��׃�����, �����ṩ���ѺõĿ�ҕ���ӿ�, ÿ�N��ͽY(ji��)��(g��u)׃������ģʽ�M(j��n)�И�(bi��o)�R, ͨ�^�ғ����(bi��o)�ɫ@ȡ�P(gu��n)���ض��x�L��Y(ji��)��(g��u)׃����������Ϣ(�D1(f)). ����֮��, inGAP-sv���Sʹ���߸���(j��)�����������`���O(sh��)���@ʾ�y������е����ͺ��B�����ɫ, �Ա���õ؞�̽���Y(ji��)��(g��u)׃���ṩ����. inGAP-svᘌ��ڽY(ji��)��(g��u)׃���ṩ�R�e����ҕ����עጡ��˹�����һվʽ�ķ���(w��), �@�N����ҕ�����ھ��һ�w, ע���Ñ��w�ȵĹ����O(sh��)Ӌ��ʽ�A(y��)ʾ��δ����ܛ���_�l(f��)����.
����2.2 ���ӽY(ji��)��(g��u)��(sh��)��(j��)
�����Y(ji��)��(g��u)��������W(xu��)�nj������ͻ��W(xu��)�c����W(xu��)���B�ӵ�һ�T�P(gu��n)�I�W(xu��)��, ����Ҫ�۽���3D��4D��(f��)�s�Π�����P(gu��n)ϵ���о�, �ɹ��(bi��o)ӛ���@�^���Լ������ȼ��g(sh��)���@һ�I(l��ng)���ṩ���S����ҕ�D��(sh��)��(j��), ���������(w��)�ڷ��ӽY(ji��)��(g��u)�Ŀ�ҕ���������о��^�������˘O�������. �Կ�ҕ��ܛ��ParaView[44]����, �����Sʹ����ͨ�^���ԺͶ����ļ��g(sh��)�ֶΌ������Ĕ�(sh��)��(j��)�����ٽ���3Dҕ�Dģ��, ������ĽǶȌ����ӽY(ji��)��(g��u)�M(j��n)���^��. ���ڵ����|(zh��)�ȴ���ӽY(ji��)��(g��u)����(x��)��(f��)�s, ���(n��i)����λ���P(gu��n)ϵ��Ҫ������Ӌ���YԴ, ���3Dҕ�Dܛ��������2Dչʾ������Ҫ���Ӹ�Ч���㷨�O(sh��)Ӌ, �����ܵ�Ӌ���O(sh��)���Լ��߷ֱ��ʵ�չʾ��Ļ. ��������(qi��ng)����Ҏ(gu��)ģ��(sh��)��(j��)����̎������, ParaViewʹ���˷ֲ�ʽ�惦Ӌ���YԴ, �����\���ڳ���Ӌ��C(j��)�ρ팦�f�|�μ��Ĕ�(sh��)��(j��)���M(j��n)�п�ҕ������. ����ParaView, Amira[45], FluoRender[46]�ȹ��߶������Á�g�[���� CT, MRI���@�D��, �Լ����F(xi��n)�����ӽY(ji��)��(g��u)��3D ߀ԭ.
�����@Щ��Ӌ��D�ΌW(xu��)����A(ch��)���_�l(f��)��ܛ�������mȻ�Ը��龫��(x��)��(zh��n)�_��չʾ��ʽȡ��������ģ��, ���Džsʧȥ���c���팦�ӽ��|�r�a(ch��n)���Ĺ��е�ҕ�X�S����, ���@�N�|�X�ͱ��w��������������3Dģ�ͺ��M(j��n)�����������ṩ���P(gu��n)�I�ľ���. ��˹��I(y��)�I(l��ng)������w��������u����(y��ng)���ڌ����ӽY(ji��)��(g��u)��߀ԭ��. Ʃ��������A��W(xu��)�c��������ِ����W(xu��)�о��ˆT�Ի���z���w�S���͌m�i����(x��)����ԭ����, �ھ���(zh��n)�ą���(sh��)������, ����һ�_3D��(x��)����ӡ�C(j��)�ɹ���������c��Ȼ�[��ʮ�ֽӽ����[��ģ��[47].
����2.3 �P(gu��n)ϵ�W(w��ng)�j(lu��)
���������I(l��ng)��������������ӻ��������x;�����{(di��o)�����úͻ�����_(d��)�ȬF(xi��n)��Ĵ��ڴ�ʹ�˸��N���ӵ��P(gu��n)ϵ�W(w��ng)�j(lu��)�Ĵ���, �S���ƌW(xu��)�҂����@Щ�^�̵������о�, �˂������(f��)�s�ȵ��˽�Ҳ�ڲ�������. ����W(xu��)�ҽ�(j��ng)����Ҫ�������������P(gu��n)ϵ�ď�(f��)�sϵ�y(t��ng)�߾S��(sh��)��(j��)�M(j��n)�з���, ��ˮa(ch��n)���˿��Ԍ����N�W(w��ng)�j(lu��)�P(gu��n)ϵ�M(j��n)�п�ҕ����ܛ������. Ŀǰ���õď�(f��)�s�W(w��ng)�j(lu��)��ҕ��������Cytoscape[53]��R�е�igraph���Լ�Perl�е� GraphViz����. Cytoscape����һ����c��ģʽ����A(ch��)�M(j��n)�оW(w��ng)�j(lu��)��ҕ���Ĺ���, ���ṩ���A(ch��)�Ĺ��ܲ��ֺ;W(w��ng)�j(lu��)��ԃ����, �����܉�����(j��)������(sh��)��(j��)�P(gu��n)ϵ�ӑB(t��i)���ɿ�ҕ���W(w��ng)�j(lu��). �������ӡ������|(zh��)�ͷ���ʹ���c��ʾ, ���c�g�Ľ����P(gu��n)ϵ���B��Ҳ����߅�M(j��n)�б�ʾ. �@�N��ʾģʽ�����˷����g����õľW(w��ng)�j(lu��), �m���κη���ϵ�y(t��ng)�ĽY(ji��)��(g��u)����P(gu��n)ϵ, ���S�������|(zh��)��DNA �������������������Ҫ���õķ��Ӕ�(sh��)��(j��)���P(gu��n)(li��n)����, �γ�����ľW(w��ng)�j(lu��)�Y(ji��)��(g��u). ����, R�е�NetBioV, Gephi[54](�D1(c))��ܛ������������Ϣ�W(xu��)���ṩ�ˌ���(ji��)�c�B����͵ľW(w��ng)�j(lu��)�P(gu��n)ϵ��ҕ���_�l(f��)����.
�����S��Ӌ���ֶε��M(j��n)һ���l(f��)չ, �W(w��ng)�j(lu��)�P(gu��n)ϵ��3D��ҕ����ʽ��u�l(f��)չ����. BioLayout Express3D[55]����������2D, 3D���g��(n��i)�Ŀ�ҕ������w�{��̽���ͷ������͵ľW(w��ng)�j(lu��)�P(gu��n)ϵ. ��ܛ���Ɍ������|(zh��)���������������Ե��P(gu��n)ϵ�γɵľW(w��ng)�j(lu��)�M(j��n)��չʾ, ���˂��y(t��ng)�Č���л�����_(d��)��(sh��)��(j��)�M(j��n)�нy(t��ng)Ӌ�W(xu��)������ķ���, �D(zhu��n)�������P(gu��n)(li��n)���u�������x���_(d��)���g��������, �Ķ��γɔ�(sh��)��(j��)�����ľW(w��ng)�j(lu��)��ʽ , ���Ҵ˹����� OpenCL���п�ܾ���, ��ֿ��]���W(w��ng)�j(lu��)�P(gu��n)ϵ3D��ҕ���r�����Ӌ���YԴ���D��̎�����g(sh��)֧�ֵȆ��}. ��2D��3D�h(hu��n)����BioLayout Express3D�ṩ����3������; (1) ���D����Ƅӡ����D(zhu��n)�Ϳs�Ų���; (2) ��(ji��)�c��߅�Ă��Ի�����, �����S�O(sh��)���ı���(bi��o)���Լӏ�(qi��ng)ʾ���; (3) �����ɫ��3D�����ͶӰ����(ji��)�c����y�����@ʾ��(n��i)�ݿ��M(j��n)��ƫ���O(sh��)��, �Ա���õ،���ҕ��Ч���M(j��n)����Ⱦ.
����2.4 �R����(sh��)��(j��)
�����mȻ��Ӳ��v��ʹ�÷����ڲ���ؔU(ku��)��, ���Dz��y(t��ng)һ�Ę�(bi��o)��(zh��n)���ǽY(ji��)��(g��u)���Ĕ�(sh��)��(j��)ģʽ���о��߫@ȡ�����ί����挍�Y������˺ܴ���ϵK. �ƌW(xu��)�҂�Ҳ�_ʼ����̎���@�����}, ����������[����(sh��)��(j��)��Ŀ��(bi��o)��Flatiron��������һ������, Flatiron��Ļ����ƶ˵�OncologyCloud[63]ƽ�_�ۺϲ��D(zhu��n)�Q�ˁ��Զ������Ļ�����Ϣ��ˎ����Ϣ�ͻ��֏�(f��)��r�Ȕ�(sh��)��(j��), ���ṩ����(sh��)��(j��)���Ěw�{����(�D1(d)), �ɴ��t(y��)�����H�܉�ͨ�^OncologyCloud����ͬ��ߵ��ί��Y(ji��)��, ߀��ۙ��������ͬ�ί��������a(ch��n)�����R���Y(ji��)��. �@��һ���ṩȫ����[����(sh��)��(j��)�ռ���������ϵ�y(t��ng)Ҳ���[���I(l��ng)��Ļ��A(ch��)�о��ṩ�˘O��ı���; “���Y�����ƌW(xu��)�f(xi��)��CEO�A����(the CEO Roundtable on Cancer)” �Ƴ���PDSӋ��(Project Data Sphere)[24], �Lԇ����һ�����Y�����R��ԇ(sh��)��(j��)�����ͷ���ƽ�_, ��(sh��)��(j��)����ِ�Z�ơ��x���Լ���˹�����șC(j��)��(g��u)��ͬ�ṩ, �@Щ��(sh��)��(j��)����ȥ�����߂�����Ϣ���M(j��n)���˽y(t��ng)һ��̖. �����f��(x��)�T��ijЩҎ(gu��)���ƶȵ�Ӱ�, �����t(y��)����(sh��)��(j��)�����Ϻ��ھ�߀��r�g���l(f��)չ��Ҏ(gu��)��. �����ɷ��J(r��n)����, ���ί���Ϣ�R����һ���M(j��n)�з���չʾ�����˼����������ɺ�ҕ������.
�������������U���Ŀ�ҕ������, ����(j��)��ͬ������߀�������ܶ������Ŀ�ҕ����ʽ (�� 1~4). �� �� , Chimera[50](�D1(b))�����ӽY(ji��)��(g��u)�Ͱ����ܶȈD�V���������b�䡢���бȌ���܉�E�ڃ�(n��i)�����P(gu��n)��(sh��)��(j��)��������, �a(ch��n)�����|(zh��)���ĄӮ�Ч��; ���ڲ�ͬ�|(zh��)�V�x���a(ch��n)���ĵ����|(zh��)�V��ʼ��(sh��)��(j��)��ʽ��ͬ, �������|(zh��)�M�W(xu��)�|(zh��)�V��(sh��)��(j��)�����нy(t��ng)Ӌ�W(xu��)�㷨�Č��F(xi��n)�^�ڏ�(f��)�s, ��(sh��)��(j��)��ʾ��ҕ����������ȡ��ҕ�������ҕ���������|(zh��)�|(zh��)�V��(sh��)��(j��)�ķ���ʮ����Ҫ; ����֮��, ߀����ᘌ���SNPչʾ�����^�z���W(xu��)���ṩ�ĺ�С�w��λ���M�������Y(ji��)���Ŀ�ҕ��������Ⱥ��śr�Ŀ�ҕ������������D����T����헌��ܵĿ�ҕ��ܛ������. �����(sh��)��(j��)��ҕ�����߷N���, ���˸��õ؞��ھ���Ч��Ϣ��䁉|, ���_�l(f��)څ������нy(t��ng)Ӌ�������ܵ�һվʽ���ɹ��߿��n. ����, δ���������(sh��)��(j��)��ҕ�������ڽ����ԡ����^�ԡ������Է��������Խ��Խ��.
����3 չ����δ��������(zh��n)
�������(sh��)��(j��)�����Լ������c, ���H��(sh��)��(j��)Ҏ(gu��)ģ����, �ֲ��ڲ�ͬ�ĽM���C(j��)��(g��u), ���ҾS�ȸ�, ��(sh��)��(j��)�������ԺͲ��_���ԏ�(qi��ng). ���ø��N���g(sh��)�ֶΫ@ȡ��(sh��)��(j��)��������Ŀ��, ����(sh��)��(j��)�M(j��n)�п�ҕ��Ҳ����Ŀ��, ������Ŀ����̽�������ı��|(zh��), �l(f��)�F(xi��n)δ֪��Ҏ(gu��)��, ����Ľ����Ҹ�����(w��), ����ھ��[���ڔ�(sh��)��(j��)����ĺ��x�ɞ�������Ϣ�W(xu��)�҂�һ�µ�Ŀ��(bi��o). ����˽�Ŀǰ�ڷ������(sh��)��(j��)�ĵ�·�ϴ��ڵ�һЩ����(zh��n)�����ڵĽ�Q����������Ҫ�����x.
��������, �F(xi��n)�еĺ������(sh��)��(j��)�д��������������������, ���a(ch��n)��(sh��)��(j��)�ĽM���C(j��)��(g��u)���Ԍ�ԭʼ��(sh��)��(j��)�M(j��n)�И�(bi��o)��(zh��n)��̎�����|(zh��)��. ����, �Ɍ���(sh��)��(j��)���T�e�, ʹ�ýy(t��ng)һ�Ĕ�(sh��)��(j��)�惦��(bi��o)��(zh��n)��Ҏ(gu��)���. �������A(y��)̎���ֶο���һ���̶��Ͻ��͔�(sh��)��(j��)Ҏ(gu��)ģ����(f��)�s��, ��(ji��)ʡ�惦���g����(sh��)��(j��)��ݔ�ɱ�, ͬ�rҲ����ߔ�(sh��)��(j��)�����x��, �p���о��ߌ���(sh��)��(j��)�M(j��n)����ͬ̎������Ҫ��Ӌ��r�g���YԴ��.
�������, ���ڮa(ch��n)���Ĕ�(sh��)��(j��)�����ֲ��ڲ�ͬ���о��C(j��)��(g��u), ��Ό��F(xi��n)������(sh��)��(j��)�Ĺ������о��ˆT���ձ����R��һ������(zh��n). �F(xi��n)�еķֲ�ʽע�ϵ�y(t��ng)(DAS)[69]�ṩ��һ�����ڵĽ�Q����. �����x��һ���Á����Q������|(zh��)���м���עጵ�ͨ�Ņf(xi��)�h, �ڴ˅f(xi��)�h��, ���ھW(w��ng)�j(lu��)�Ŀ�ҕ��ϵ�y(t��ng)�Ɍ��F(xi��n)ͬһ�������h(yu��n)�̮��طֲ�עጔ�(sh��)��(j��)�Ŀ�ҕ��.
��������, ���(sh��)��(j��)���еď�(f��)�s�����Խo��(sh��)��(j��)�ھ��ܴ����y, ����ڌ���������(sh��)��(j��)�M(j��n)�п�ҕ��ǰ, ��(sh��)��(j��)ͶӰ�����N���;S�ȵļ��g(sh��)���V������. �c��ͬ�r, ���ҕ�X�����J�ԡ�ʹ�����挦չʾ����r���Ɣ���������Ϣ�������������ض���Ҫ���Կ��]. �������(sh��)��(j��)�M(j��n)�п�ҕ���r, ��ҪӛסĿ��(bi��o)ʹ��������, Ŀ������Ϣ��չʾ��̽��, ����һζ����ҕ�X���^. ���_�l(f��)�����(sh��)��(j��)�Ŀ�ҕ�����ߕr, ��Ҫ�M�������ܛ����ƽ�_��������, ��ֿ��]�Ñ����w��, �ṩ�ѺõĽ�������.
��������, �����ĕr�g��(n��i)����Ҏ(gu��)ģ��(sh��)��(j��)�M(j��n)��̎������ҕ�����������Ҫ��. ����ͨ�^ʹ�Ã�(y��u)���㷨����(sh��)��(j��)Ҏ(gu��)ģ�Ϳ�ҕ��Ч���M(j��n)��ƽ����, ߀�������벢��̎�����g(sh��). �ڌ����ɔ�(sh��)��(j��)���M(j��n)�п�ҕ���r, �Ɍ���ԃ̎����ɢ�ڶ������й�(ji��)�c��, �Դ˿s���\�Еr�g, �ӿ��ҕ�����ٶ�.
��������ǰ����(n��i)��, ���ڂ�ݔ���(sh��)��(j��)�ľW(w��ng)�j(lu��)���A(ch��)�O(sh��)ʩ�Ľ��O(sh��)����(sh��)��(j��)�Ĵ惦��ʽ���T��涼������һ�������y. �mȻ�ڷ��������(sh��)��(j��)�ĵ�·�����R���T������(zh��n), �����@Щ���r�����y��������ֹ�ƌW(xu��)�҂�ǰ�M(j��n)���_��, �����ƌW(xu��)�������漆��K������һ���������ˆT��Ŭ���±���ȫ���_.——Փ�����ߣ����բ� , ���ע� , �w���c��*